2 Теоретическая часть
Характеристики удерживания
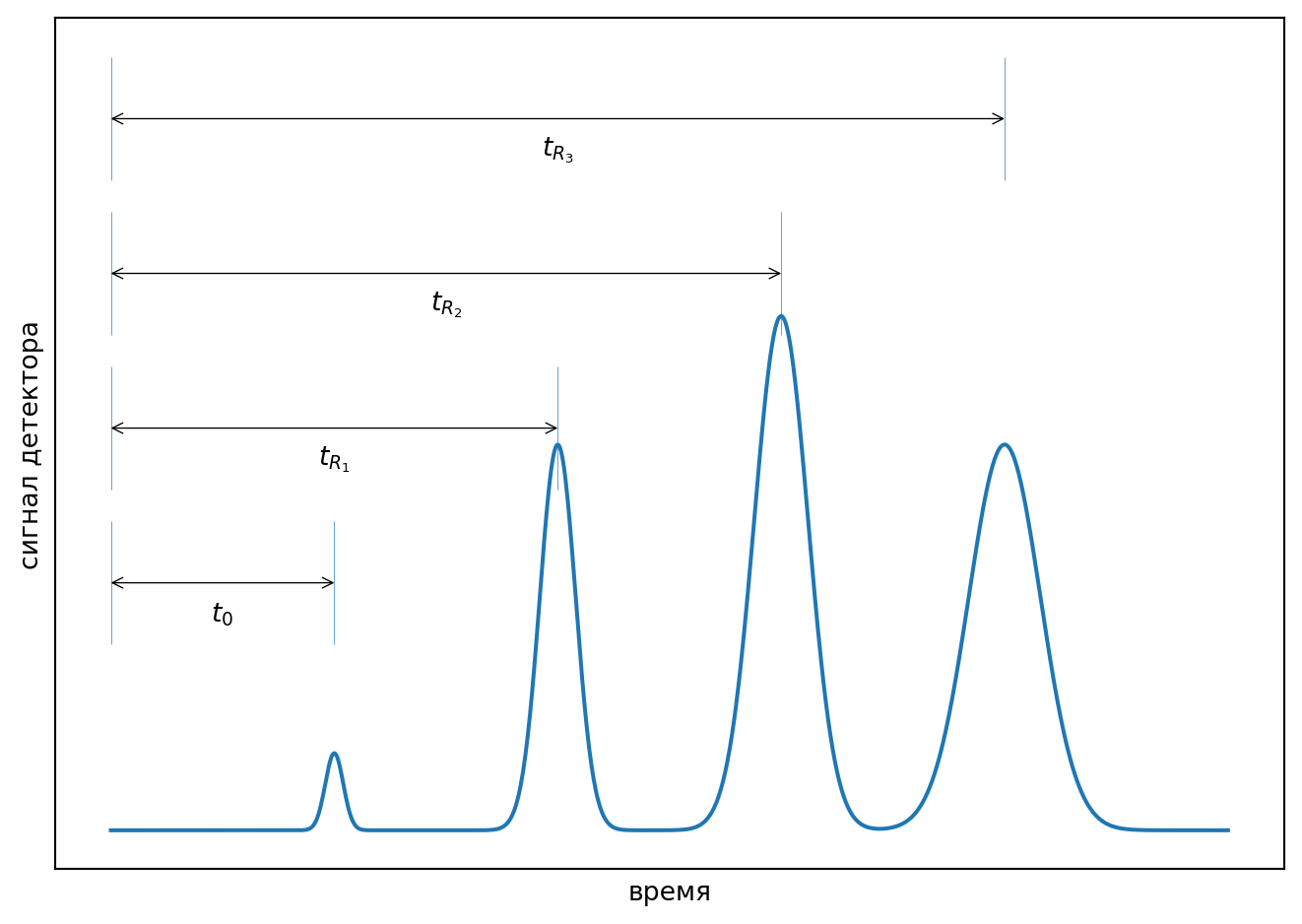
В газовой хроматографии источником информации для качественного и количественного определения анализируемой смеси является хроматограмма (рис. 2.1).
Непосредственно измеряемой величиной в газовой хроматографии является время удерживания данного компонента \(t_R\), измеряемое обычно в минутах, т.е. время, протекающее от момента ввода вещества в колонку до момента его выхода из колонки, фиксирусмого детектором. Если полоса размывается за счет продольной диффузии, то обычно время удерживания отсчитывается до выхода максимума хроматографической полосы (рис. 2.1).
Удерживание в газохроматографической колонке определяется взаимодействием компонентов пробы с сорбентом (или, в случае распределительной хроматографии, — с неподвижной жидкой фазой), а также временем прохождения вещества по газовой линии прибора от места введения пробы до чувствительного элемента детектора.
Время прохождения вещества по газовой линии определяется по времени выхода несорбирующегося газа — \(t_0\) (мёртвое время). Удерживание несорбирующегося газа зависит от состояния газовой линии прибора. Если в колонке свободный (мертвый) объём \(V_M\) (мл) мал, то движение зоны несорбирующегося вещества идет быстрее и наоборот, поскольку \[ t_0 = V_M/F = L/u. \] Здесь и далее \(F\) — объемная скорость газа (мл/мин), \(L\) — длина колонки (см), а \(u\) — линейная скорость газа (обычно выражают в см/с).
Мёртвое время определяют обычно следующим образом: если хроматограф работает с детектором по теплопроводности и газ-носитель гелий или водород, тогда на хроматограммме при введении пробы появляется пик воздуха, который отмечает время выхода пика несорбирующегося газа (пик воздуха появляется потому, что при проколе мембраны в прибор вносится его некоторое количество).
Если прибор работает с детектором по теплопроводности и газ-носитель азот, то пик воздуха не появляется из-за близкой теплопроводности воздуха и азота. В этом случае в прибор вводят некоторое количество гелия (или водорода), пик которого соответствует удерживанию не сорбирующегося газа.
Если работать с пламенно-ионизационным детектором, место пика на хроматограмме не сорбирующегося газа можно найти, вводя в колонку метан, который относится к практически не сорбирующимся газам.
Исправленное время удерживания вещества представляет собой разность \[ tR' = t_R - t_0. \]
По своему смыслу исправленное время удерживания — это часть общего времени удерживания, в течение которой вещество удерживалось неподвижной фазой. Исправленное время удерживания по-прежнему зависит от геометрических параметров колонок.
Величина \[ k = t'_R/t_0 = (t_R-t_0)/t_0 = t_R/t_0 - 1 \] называется фактором ёмкости колонки к определяемому веществу. Эта величина не зависит от геометрических параметров колонки (длина и диаметр), однако зависит от соотношения объемов доступного для газа пространства колонки (\(V_M\)) и объёма НЖФ \(V_S\) (в распределительном варианте хроматографии) или удельной поверхности сорбента (в адсорбционном варианте).
Проще всего показать это на примере распределительной хроматографии. Время удерживания вещества и объёмная скорость потока \(F\) определяют удерживаемый объём газа \(V_R\), который требуется пропустить через колонку для выхода вещества: \[ V_R = Ft_R. \]
Часть этого объёма занимает газ, вытесненный из колонки: \[ V_M = Ft_0. \]
Таким образом, исправленный удерживаемый объём газа — это именно тот объём газа, который находился в равновесии с объёмом НЖФ \(V_S\) в процессе хроматографирования вещества: \[ V'_R = V_R-V_M = F(t_R-t_0). \]
Фактор ёмкости \(k\), таким образом, также можно выразить через объёмы газов: \[ k = \frac{t'_R}{t_0} = \frac{Ft'_R}{Ft_0} = \frac{V'_R}{V_M}. \tag{2.1}\]
Концентрация вещества в газовой фазе \(c_M\) и в НЖФ \(c_S\) связаны безразмерной константой распределения вещества между фазами: \[ K_D = c_S/c_M. \]
Для хроматографической колонки, однако, удобнее записать отношение не концентраций, а количеств (моль, г) веществ в различных фазах, поскольку объёмы \(V_M\) и \(V_S\) для колонки фиксированы и полученная величина будет для колонки также константой: \[ \frac{\text{кол-во в-ва в объёме НЖФ}}{\text{кол-во в-ва в объёме газа $V_M$}} = \frac{c_SV_S}{c_MV_M} = \frac{K_DV_S}{V_M} = const \] Но произведение \(K_DV_S\) имеет размерность объёма, и, таким образом, мы получили в правой части то же отношение, что и в уравнении (2.1), то есть: \[ V'_R = K_DV_S. \tag{2.2}\] Исправленный удерживаемый объём, таким образом, определяется объёмом НЖФ в колонке. Отношение же, которое мы получили — значение фактора ёмкости колонки, записанное через коэффициент распределения вещества и отношения объёмов НЖФ и газовой фазы в колонке.
Величина \[ \beta = \frac{V_M}{V_S} \] называется фазовым отношением и характеризует степень заполнение колонки НЖФ.
Таким образом, окончательно получаем: \[ k = \frac{t_R-t_0}{t_0}= \frac{V'_R}{V_M} = K_D\frac{V_S}{V_M} = K_D\frac{1}{\beta}. \tag{2.3}\]
В случае газоадсорбционного варианта хроматографии в роли объёма НЖФ \(V_S\) выступает величина удельной поверхности сорбента.
Фактор ёмкости \(k\), таким образом, — основная характеристика удерживания вещества (сорбата) НЖФ (или сорбентом) — определяется двумя параметрами:
- Термодинамической константой распределения (адсорбционного равновесия) вещества между фазами. Эта величина характеризует взаимодействие разделяемого вещества и неподвижной фазы и определяется их природой. Константа распределения экспоненциально зависит от температуры, что позволяет использовать температуру колонки для управления удерживанием веществ в широких пределах.
- Количеством НЖФ в колонке (толщиной плёнки НЖФ) или удельной поверхностью сорбента. Фазовое отношение \(\beta\) колонки можно менять в случае ГЖХ в несколько раз, выбирая фазы с различной долей НЖФ на носителе. Для анализа летучих веществ выбирают колонки с максимальным содержанием НЖФ на носителе, для нелетучих — наоборот, с минимальным.
Связь формы изотермы распределения вещества с формой пика
Для линейной изотермы распределения (адсорбционного равновесия) \(K_D = dc_S/dc_M = const\) величина \(k\) не зависит от концентрации вещества в подвижной фазе, регистрируемый пик симметричен. Такая форма пика наблюдается в случае ввода в колонку очень малых количеств веществ.
Для выпуклой изотермы распределения Лэнгмюровского типа характерно уменьшение наклона \(dc_S/dc_M\) с ростом концентрации вещества в подвижной фазе \(c_M\). Это приводит к тому, что в области высоких концентраций вещества в подвижной фазе удерживание уменьшается — верхушка пика движется быстрее, а пик становится асимметричным, причём правый край пика (тыл) становится более пологим. Большинство пиков в газовой хроматогафии имеют именно такой вид асимметрии.
Наоборот, для вогнутой изотермы распределения, характерной для полимолекулярной адсорбции, с ростом концентрации вещества в подвижной фазе наклон \(dc_S/dc_M\) увеличивается, что приводит к замедлению скорости движения верхушки пика — пик становится асимметричным, причём левый край пика (фронт)становится более пологим. Такие пики наблюдаются в газовой хроматографии при низких температурах колонки, когда пары вещества конденсируются в плёнку на поверхности сорбента.
Объёмные характеристики скорости потока
Объемная скорость газа-носителя обычно измеряется на выходе хроматографической колонки. Наиболее простым и надёжным способом измерения является пенный измеритель расхода, который представляет собой бюретку с резервуаром (резиновой грушей), заполненным мыльным раствором. Через бюретку проходит газ-носитель и захватывает мыльную пленку. С помощью секундомера определяют время, за которое мыльная пленка проходит расстояние между двумя калибровочными метками на бюретке, и рассчитывают объемную скорость потока.
Измеряемая скорость газа-носителя в пенном измерителе не представляет истинную объемную скорость в колонке, т.к. измерение скорости проводится при комнатной температуре, а колонка находится при температуре значительно более высокой. Кроме того, надо иметь ввиду, что мыльный раствор в пенном измерителе дает определенное для данной температуры давление водяных паров. Чтобы привести определяемую скорость \(F_\text{изм.}\) к истинной скорости \(F_o\), необходимо ввести поправку на температуру колонки и на давление водяных паров в пенном измерителе: \[ F_o = F_\text{изм.}\frac{T_\text{c}}{T_\text{комн}}\frac{p_o-p_w}{p_o}. \] где \(T_c\) — температура колонки, K; \(T_\text{комн}\) — температура пенного измерителя (обычно принимается равной комнатной, K); \(p_o\) — давление на выходе колонки (обычно атмосферное); \(p_w\) — давление водяных паров при температуре пенного измерителя. Множитель, дающий поправку на давление водяных паров, имеет величину очень близкую к 1, и поэтому его вычисляют только при очень точных измерениях.
Зёрна неподвижной фазы хроматографической колонки представляют сопротивление на пути газового потока. Для того, чтобы протекал газ, давление на входе должно быть выше, чем на выходе колонки. По длине колонки устанавливается градиент давления, который приводит к разной объемной скорости в каждом участке колонки. Из-за сжимаемости газа-носителя объемная скорость на входе в колонку меньше, чем на выходе, поэтому вводится специальная поправка \(j\), учитывающая перепад давления, для определения средней скорости газа, известный как коэффициент Джеймса–Мартина \(j\): \[ j = \frac{3(P^2-1)}{2(P^3-1)}, \] где \(P=p_i/p_o\) — отношение давления на вход в колонку \(p_i\) к давлению на выходе колонки \(p_o\) (обычно равного атмосферному). Входное давление измеряется по манометру (то есть относительно атмосферного) в кг/см2, а атмосферное — часто в мм рт. ст. (1 кг/см2 = 735.56 мм рт.ст.). Для расчёта поправки входное давление необходимо пересчитать в абсолютные величины!
Например, для случая, если барометрическое давление равно 750 мм рт.ст., а манометр на входе в колонку показывает 1.5 кг/см2: \[ P = \frac{p_i}{p_o} = \frac{p_\text{ман.}+p_\text{бар.}}{p_\text{бар.}} = \frac{1.5\times735.56+750}{750} = 2.471. \] \[ j = \frac{3(P^2-1)}{2(P^3-1)} = \frac{3(2.471^2-1)}{2(2.471^3-1)} = 0.5436. \]
Объем удерживания \(V_R\), как было показано выше, вычисляется как произведение времени удерживания на объёмную скорость элюента. Когда в величину объемной скорости удерживания введены указанные поправки, получается так называемый эффективный или истинный объем удерживания \(V_N\): \[ V_N = jV'_R = jF_ot'_R. \]
Удерживаемый объем, как следует из уравнения (2.3), зависит от количества твердого адсорбента в колонке в случае ГАХ или от количества НЖФ в ГЖХ, поэтому для физико-химических измерений используют понятие удельного удерживаемого объема \(V_g\) — это эффективный удерживаемый объем, относящийся к 1 г сорбента (НЖФ) и прведённый к нормальным условиям, т.е.: \[ V_g = \frac{V_N}{m_S}\frac{273.15}{T_c}\frac{p_o}{p_n}, \tag{2.4}\] где \(m_S\) — масса сорбента (НЖФ), г; \(p_n\) — нормальное атмосферное давление (101.325 кПа).
В случае ГЖХ объём НЖФ равен отношению массы жидкой фазы к ее плотности, и поэтому из уравнений (2.2) и (5.7) имеем \[ V_g= K_DV_S/m_S = K_D/\rho. \]
Поскольку \(K_D\) и \(\rho\) при постоянной температуре определяются только природой системы растворяющийся компонент—НЖФ, то удельный удерживаемый объём в газо-жидкостной хроматографии обладает свойствами физико-химической константы. В справочные данные поэтому табулируются именно величины \(V_g\).
В газоадсорбционной хроматографии объем адсорбционного слоя определяется площадью поверхности адсорбента \(S\) и его толщиной \(\delta\): \[ V'_R = S\delta = S_\text{уд.}m_S\delta, \] где \(S_\text{уд.} = S/m_S\) — удельная поверхность адсорбента (м2/г).
С учетом этого удельный удерживаемый объём \[ V_g = S_\text{уд.}\delta K. \]
Так как адсорбент может обладать различной удельной поверхностью, то величина удельного удерживаемого объема в случае ГАХ не определяется только природой системы адсорбат–адсорбент, т.е. не является физико-химической константой. Такой константой будет величина абсолютного удерживаемого объема \(V_a\), т.е. величина удельного удерживаемого объема, отнесенная к удельной поверхности твердого адсорбента: \[ V_a = V_g/S_\text{уд.}. \]
Величина \(V_a\) не зависит от площади для однородной поверхности, поэтому для твердых тел одинаковой природы (но разной дисперсности) можно легко найти сравнительным методом их удельную поверхность: \[ S_\text{уд.} = \frac{V_g}{V_{g,\text{ст.}}}S_\text{уд.ст.}. \]
В лабораторной практике часто используются относительные величины удерживания, которые дают более высокую точность при идентификации хроматографических пиков.
Для получения относительных характеристик удерживания выбирают исправленные объемы удерживания или исправленные времена удерживания определяемых веществ и относят эти величины к соответствующим величинам одного из веществ (чаще всего \(н\)-алкана), принятым за стандарт.
Относительный объем удерживания зависит от природы используемого сорбента и температуры опыта. Остальные экспериментальные параметры, такие как скорость газового потока, масса жидкой фазы, величина поверхности адсорбента, давление на входе и выходе колонки не влияют на его величину. Поэтому ошибки в этих параметрах не сказываются на величинах относительных объемов удерживания.
Эффективность хроматографической колонки
Для оценки эффективности работы хроматографической колонки, по аналогии с ректификацией, применяют высоту теоретической тарелки \(H\) («высота, эквивалентная теоретической тарелке», сокращенно ВЭТТ), которая определяется как длина колонки \(L\), деленная на число теоретических тарелок \(N\). Число теоретических тарелок показывает, сколько раз должно устанавливаться равновесие между газовой и неподвижной фазами в идеальном ступенчатом процессе, эквивалентном результатам работы реальной колонки. Чем больше число этих ступеней (тарелок), т.е. чем меньше ВЭТТ, тем больше эффективность колонки.
<>:26: SyntaxWarning:
invalid escape sequence '\s'
<>:28: SyntaxWarning:
invalid escape sequence '\s'
<>:36: SyntaxWarning:
invalid escape sequence '\s'
<>:46: SyntaxWarning:
invalid escape sequence '\s'
<>:26: SyntaxWarning:
invalid escape sequence '\s'
<>:28: SyntaxWarning:
invalid escape sequence '\s'
<>:36: SyntaxWarning:
invalid escape sequence '\s'
<>:46: SyntaxWarning:
invalid escape sequence '\s'
/var/folders/gy/s_tn_jqd225c2r7mv1236gj00000gn/T/ipykernel_12829/2543929579.py:26: SyntaxWarning:
invalid escape sequence '\s'
/var/folders/gy/s_tn_jqd225c2r7mv1236gj00000gn/T/ipykernel_12829/2543929579.py:28: SyntaxWarning:
invalid escape sequence '\s'
/var/folders/gy/s_tn_jqd225c2r7mv1236gj00000gn/T/ipykernel_12829/2543929579.py:36: SyntaxWarning:
invalid escape sequence '\s'
/var/folders/gy/s_tn_jqd225c2r7mv1236gj00000gn/T/ipykernel_12829/2543929579.py:46: SyntaxWarning:
invalid escape sequence '\s'
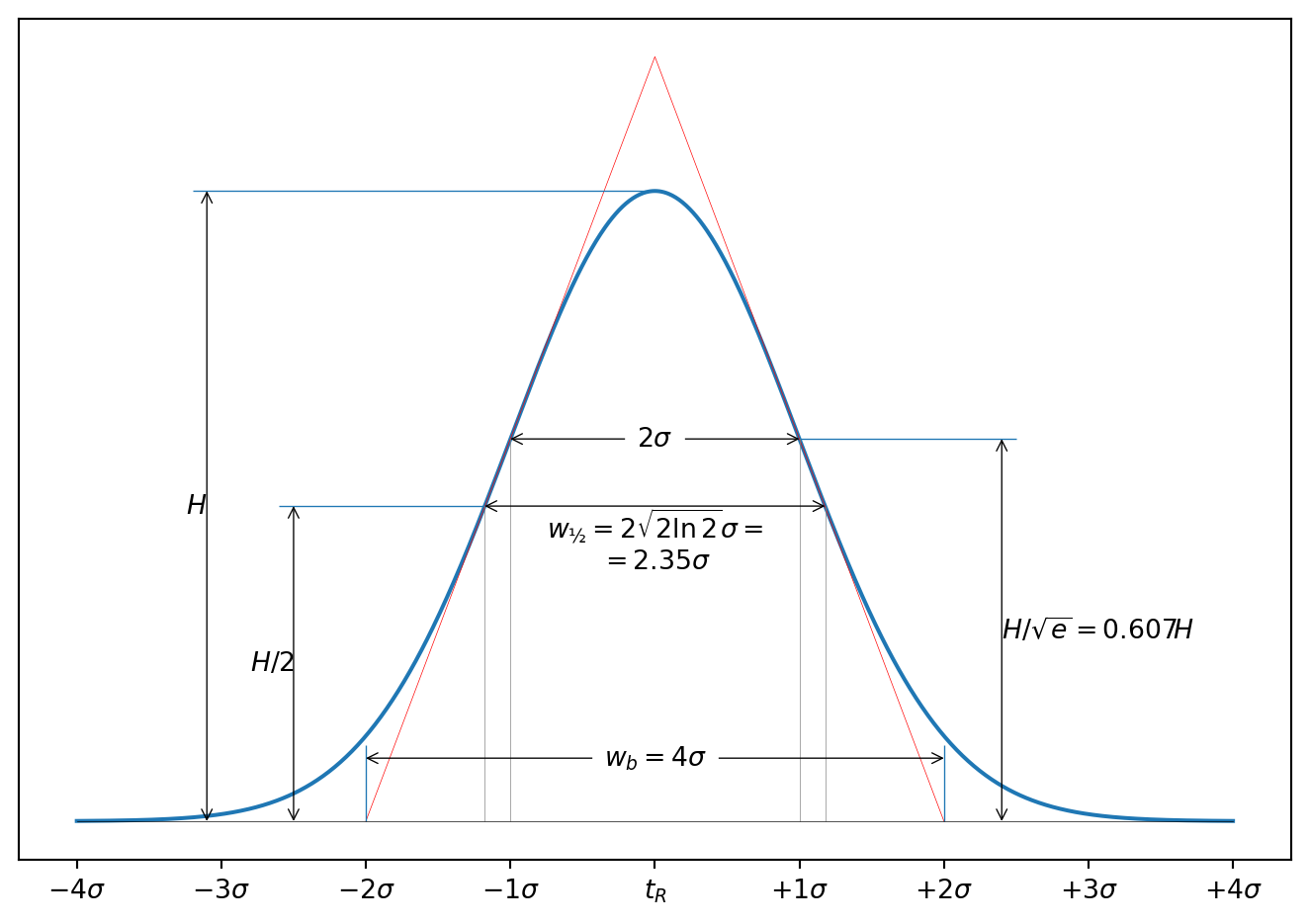
В случае линейной хроматографии форма хроматографического пика близка к гауссовой: \[ y = \frac{1}{\sigma\sqrt{2\pi}}e^\frac{(t-t_R)^2}{2\sigma^2}. \tag{2.5}\] В этом случае параметр \(\sigma\) — суммарная дисперсия, характеризующая вклад отдельных факторов в размывание пика и связанная с \(N\) соотношением: \[ N = \left(\frac{t_R}{\sigma}\right)^2. \tag{2.6}\]
Число теоретических тарелок \(N\) легко определить из хроматограммы по ширине пика, которую можно измерить на различной высоте:
На пересечении касательных в точках перегиба хроматографического пика, которые лежат примерно на 0.6 высоты пика, с базовой линией: \[ N = 16\left(\frac{t_R}{w_b}\right)^2. \]
На ширине пика, рассчитанной на на половине его высоты: \[ N = 5.54\left(\frac{t_R}{w_½}\right)^2. \]
Величина Н (ВЭТТ) позволяет проводить сравнение колонок различной длины и устанавливать меру эффективности хроматографической колонки.
С практической точки зрения также важно, что высота пика, соответствующая максимуму гауссовой функции при \(t=t_R\) (2.5), понижается с размыванием и увеличением ВЭТТ: \[ H \propto \frac{1}{\sigma}. \]
После подстановки \(\sigma\) из уравнения (2.6) получаем: \[ H \propto \frac{1}{\sigma} \propto \frac{\sqrt{N}}{t_R}. \] То есть высота пика пропорциональна \(\sqrt{N}\) и обратно пропорциональна времени выхода вещества из колонки. Для колонок с небольшим числом \(N\) (насадочные колонки имеют эффективность до нескольких тысяч теоретических тарелок, что немного) это приводит к сильному размыванию поздних пиков на хроматограмме и существенному ухудшению возможности количественного определения этих веществ.
Уравнение Ван-Деемтера
Эффективность хроматографического разделения зависит от линейной скорости газа-носителя. Эта зависимость описывается уравнением Ван-Деемтера \[ H = A + B/u + Cu, \] где \(Н\) — ВЭТТ, \(A\) — слагаемое, учитывающее процессы омывания зерен неподвижной фазы, \(B\) — слагаемое, отражающее влияние продольной молекулярной диффузии, \(C\) — коэффициент, учитывающий задержку массообмена между газом и неподвижной фазой, \(u\) — линейная скорость газа-носителя.
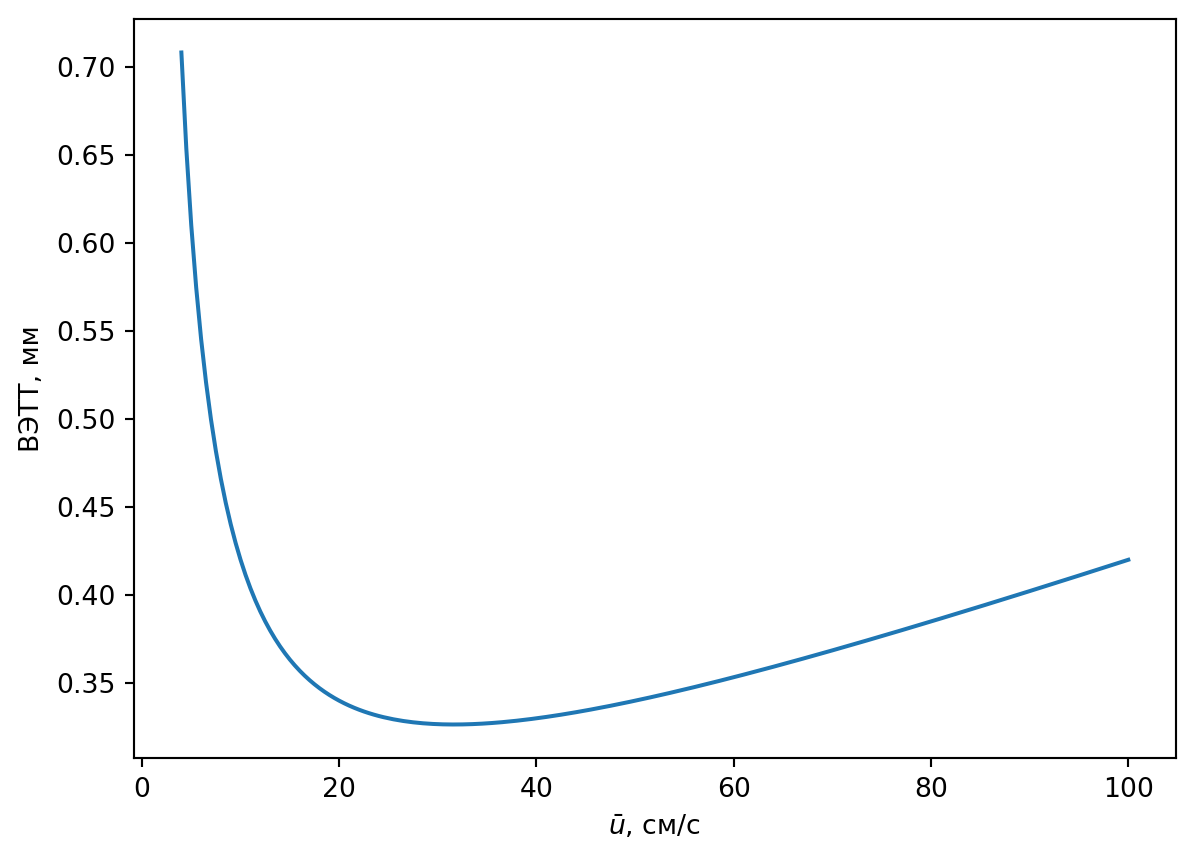
При малых скоростях потока размывание пиков, затрудняющее разделение, происходит главным образом за счёт продольной диффузии; при больших скоростях — за счёт запаздывания процессов внутренней диффузии в порах зерен и массообмена с неподвижной фазой.
На рис. 2.3 видно, что зависимость \(H\) от \(u\) (или \(F\)) имеет минимум. Следовательно, имеется некоторая оптимальная скорость газа-носителя, при которой ВЭТТ минимальна и можно достичь наилучшей эффективности колонки. Поэтому прежде чем приступить к серийным измерениям, подбирают с помощью уравнения Ван-Деемтера оптимальную для данной колонки скорость газа. Для этого необходимо снять несколько хроматограмм одного или нескольких веществ при разных скоростях газа-носителя, рассчитать \(N\) и \(H\), построить график зависимости ВЭТТ от скорости потока газа, из которого определить \(F\).